

Mutations are deviations from the normal genetic “blueprint” that is defined in the genetic material (DNA) of a cell. Certain mutations can cause a cell to become malignant and thus cause cancer, as is the case in GIST. Special testing to identify mutations found in tumor cells is classically done by determining the nucleotide sequence of a certain gene. Although mutation testing has been largely a research tool so far, it is becoming more and more available at pathology laboratories. Mutational analysis is especially important in GIST because most GISTs are defined by activating mutations in the KIT or PDGFRA genes. Moreover, the response of GISTs to certain drug treatments varies according to the mutation status of the tumors. It is therefore likely that mutational testing will become routine for GIST samples in the near future.
For a list of labs that perform mutation testing on GISTs, see our page Mutation Testing.
Historically, Hirota et al (1998) revealed not only that most GISTs are positive for the KIT protein by immunohistochemical testing, but also that most GISTs show mutations of the KIT gene. This gene is responsible for encoding the production of the KIT protein by cells. Later on it was noted by Heinrich et al (2003) that GISTs can also contain activating mutations of the PDGFRA gene.
KIT and PDGFRA Structure and Function
Both KIT and PDGFRA proteins are receptors for growth factors that are activated in normal tissues only in certain situations, for example when new cells are needed. In the normal setting, the signal that tells a cell to proliferate (reproduce by cell division) is conveyed by the appropriate ligand that binds to the extracellular portion of a receptor. This binding brings two receptors of the same kind together (dimerization, or more precisely homodimerization). The relevant ligands for the KIT and PDGFRA receptors are stem cell factor and PDGF-AA, respectively. Following ligand binding, the intracellular portions of the receptor are activated, triggering cell pathways that up-regulate proliferation, down-regulate apoptosis (programmed cell death), and control cell differentiation, adhesion, and motility (see Figure 7). Mutations of KIT and PDGFRA lead to independent activation of the receptor in the absence of ligand; this is called constitutive activation. This means the cell is carrying out spontaneous proliferation without the normal signaling ligand telling the cell to grow. The result is uncontrolled growth of a tumor. The “downstream” pathways that come into play in the cell following activation of mutant KIT or mutant PDGFRA are thought to be fairly similar (Heinrich et al, 2003 in Science; Duensing et al , 2004 in Oncogene). However, different sets of genes are expressed depending on which gene is mutant (Antonescu et al, 2004; Subramanian et al, 2004; Kang et al, 2005).
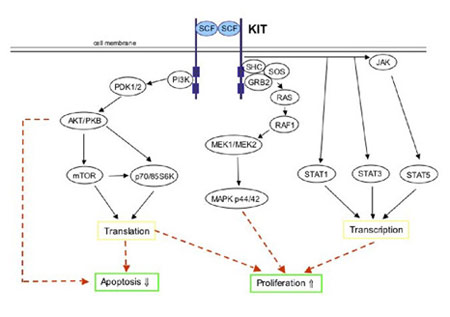
Figure 7. Simplified schematic of KIT signal transduction. Physiologic KIT activation is triggered by binding of the dimeric KIT ligand stem cell factor (SCF) leading to homodimerization of two KIT molecules. This is accompanied by structural changes in the receptor resulting in activation of the KIT kinase domain. Phosphorylated tyrosine residues on KIT serve as binding sites for various cell signaling proteins. These proteins include members of the PI3-K/AKT, RAS/RAF/MAPK and the JAK/STAT pathways. Ultimately, the phosphorylated KIT receptor stimulates intracellular signaling pathways controlling cell proliferation, adhesion, apoptosis, survival and differentiation. (Figure graciously provided by Anette Duensing, M.D.)
The two receptors KIT and PDGFRA have a similar structure, as they belong to the same sub-family of proteins within the family of receptor tyrosine kinases. A diagram of the structure of the KIT and PDGFRA receptors is shown as Figure 8. KIT and PDGFRA are comprised of an extracellular domain (the portion extending outside of the cell membrane), a juxtamembrane domain (the portion just inside of the cell membrane), and two sections inside the cell known as the two catalytic parts of the kinase domain. One part of the kinase domain is essential for the binding of ATP and the other part is needed for the transfer of a phosphate group that leads to kinase activation.
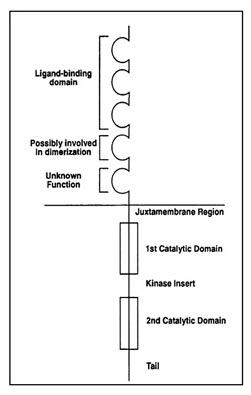
Figure 8. Structure of the receptor tyrosine kinases KIT and PDGFRA. (Figure graciously provided by Anette Duensing. M.D.)
Exon 9 of KIT codes for part of the extracellular region of KIT that is involved in receptor dimerization. KIT exon 11 and PDGFRA exon 12, respectively, code for the juxtamembrane regions of these proteins. The ATP binding domains of KIT and PDGFRA are coded for by exons 13 and 14, respectively, whereas exons 17 and 18 code for the phosphotransferase domains of these respective proteins.
KIT and PDGFRA mutations in GISTs
Over the last few years many different mutations have been detected in the KIT gene. Not only can the different mutations be found in different regions (eons) of the KIT gene, but also mutations of a single exon can consist of different structural changes in the DNA. Specifically, one can find so-called point mutations, deletions and insertions. A point mutation can be explained by the fact that one nucleotide is exchanged by another nucleotide. For example, in place of an adenosine one can now find a guanine (A -> G). Deletion mutations are characterized by the fact that some of the genetic material is lost. This can be just one, or several, or more than a hundred missing base pairs. Insertion mutations result in some nucleotides being gained, or added in a place where they do not belong. A common theme of all the KIT mutations in GISTs is that they are “in-frame”, meaning that the reading frame of three nucleotides coding for one amino acid is not changed. Consequently, the difference between a normal KIT protein and a mutated KIT protein consists only of a slightly different protein structure.
As mentioned above, KIT mutations can be found in different sections of the KIT gene. On the DNA level, genes are made up of special regions, called introns and exons. Exons are the regions of DNA within a gene that are not spliced out from the transcribed RNA and are retained in the final messenger RNA. The exons therefore contain the final coding sequences for the final protein. Mutations in the KIT gene that are relevant for GISTs are found in exons 9, 11, 13 and 17. These regions of the KIT gene correspond to different regions of the KIT protein, as shown in Figures 7 and 9. Recently a family with a mutation in exon 8 of KIT has been described (Hartmann et al, 2005), as well as two sporadic (nonfamilial) cases with this same exon 8 mutation, deletion p.Ddel419 (Huss et al, 2013). Therefore, exon 8 mutations may account for a very small number of GISTs.
Mutations in KIT can be found in about 80% of GISTs. Even GISTs that are negative or very weak for KIT immunoreactivity can nevertheless harbor KIT mutations. However, about 8% of GISTs have a normal KIT gene (wildtype) but show mutations in the gene for PDGFRA (Heinr
ich et al, 2003; Heinrich et al, 2003; Hirota et al, 2003). Most of them are negative or weakly positive for KIT expression (Medeiros et al, 2004; Corless et al, 2005; Pauls et al, 2005). PDGFRA is a protein that is member of the same family as the KIT protein. KIT and PDGFRA are both receptor tyrosine kinases and have a very similar structure (see Figure 8). The PDGFRA gene is also very similar to the KIT gene, and PDGFRA mutations have been found in exons 12, 14 and 18 (corresponding to exons 11, 13 and 17 of KIT). Interestingly, the genes coding for KIT and PDGFRA receptors are found in close proximity on chromosome 4q12 (Spritz et al, 1994). Figure 9 illustrates the proportions of GISTs with mutations of the exons coding for various regions of the KIT and PDGFRA proteins.
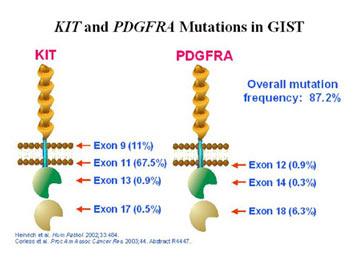
Figure 9. Diagrammatic representation of the structure of the KIT and PDGFRA receptors showing the proportion of GISTs with mutations identified in each exon. (Figure graciously provided by Cristina Antonescu, M.D.)
A common feature of mutations in KIT and in the gene for PDGFRA is that they are oncogenic, meaning that they initiate the cancer and drive the tumor. That is, either KIT mutations or PDGFRA mutations may cause GISTs. A few GISTs that are wildtype (normal) for both KIT and PDGFRA have some other (as yet unidentified) oncogenic mechanism. Researchers continue to search for causal mutations. As discussed below, nearly all GISTs in children and in NF1 patients are wildtype.
Clinical Characteristics by Mutation Type
As mentioned above, GISTs may develop either after KIT mutation or PDGFRA mutation, or sometimes without any known mutation (wildtype). Interestingly, tumors with different mutations do show differing clinical characteristics. In a series of 322 GISTs (Corless, Fletcher & Heinrich, 2004), 80.9% showed KIT mutations, while 7.1% showed PDGFRA mutations, and 12% were wildtype. The KIT mutant GISTs are 95% KIT-positive. The majority have mutations in exon 11 (66.1%) and show spindle cell morphology. Exon 11 is the most common mutation for GISTs in all anatomical locations. About 13% of KIT-mutant GISTs show exon 9 mutations (Corless, Fletcher & Heinrich, 2004). These are usually located in the small intestines and are more likely to show epithelioid or mixed morphology (Antonescu et al, 2003; Corless, Fletcher & Heinrich, 2004). Mutations in exons 13 and 17 of KIT each occur in around 1% of GISTs or less (Lux et al, 2000; Rubin et al, 2001; Corless, Fletcher & Heinrich, 2004). Epithelioid or mixed cell-type GISTs from non-gastric locations usually show KIT mutations, but gastric epithelioid GISTs are most likely to show PDGFRA mutations or no mutations (Wasag et al, 2004; Wardelmann et al, 2004). Although most tumors display only a single mutation, there are a few reports of multiple mutations within the same gene of a single tumor (Sakurai et al, 2001; Miettinen & Lasota, 2003; Kitamura & Hirota, 2004; Emile et al, 2004; Vu et al, 2005). However, mutations of KIT and of PDGFRA are mutually exclusive in primary, untreated GISTs.
The PDGFRA-mutant GISTs are nearly always of epithelioid or mixed cell types, but about 8% are of spindle morphology (Lasota et al, 2004, Medeiros et al , 2004; Penzel R et al , 2005). A common feature seems to be the occurrence of multinucleated giant cells (Pauls et al, 2005). The PDGFRA-mutant GISTs most often arise from the stomach (Corless, Fletcher & Heinrich, 2004; Wasag et al, 2004; Wardelmann et al, 2004). Most PDGFRA-mutant GISTs have low expression levels of the KIT protein as assessed by immunohistochemistry (Penzel et al, 2005).
Pauls et al (2005) reported that immunohistochemical staining patterns for KIT and PDGFRA may predict the mutation type prior to performing more expensive and time-consuming mutation analyses. Interestingly, they found that most GISTs stained for both KIT and PDGFRA. However, the staining intensity strongly correlated with the mutation type: GISTs with KIT mutations would stain strongly for KIT and weakly for PDGFRA and vice versa. Rossi et al (2005) also successfully used immunohistochemical tests to detect reactivity for PDGFRA although they did not correlate their results with the mutation type.
Adult sporadic GISTs that are wildtype for both KIT and PDGFRA may be either spindle (57%) or epithelioid type (Wasag et al, 2004; Medeiros et al, 2004; Pauls et al, 2005). Pediatric KIT wildtype GISTs are often epithelioid. GISTs occurring in patients with NF1 are often KIT wildtype and are usually of the spindle cell type.
Drug Response Varies by Mutation Type
Recent data indicate that the response of GIST patients to tyrosine kinase inhibitors (drugs including Gleevec and Sutent) varies by the specific mutation displayed by their tumors. Published work (Heinrich, Corless, Demetri et al, 2003; Corless, Fletcher & Heinrich, 2004; Debiec-Rychter et al, 2004) and data presented at the 2005 meeting of the American Society for Clinical Oncology (ASCO) indicate a stronger and longer-duration response to Gleevec for patients with KIT mutations in exon 11 than for those with mutations in exon 9 (Heinrich et al, 2005, ASCO) or for patients with GIST negative for KIT expression (Blackstein et al, 2005, ASCO). In contrast, response to Sutent in patients who had grown resistant to Gleevec was better for patients with KIT exon 9 mutations (Demetri et al, 2005, ASCO). Corless et al (2005) report data derived from cell culture models indicating that response to Gleevec in tumor cells with mutant PDGFRA depends on the particular mutation involved (also see Heinrich et al , 2003 in J of Clinical Oncology). They predict that about 35% of patients with PDGFRA mutations will benefit from Gleevec.
Second Mutations in Drug Resistance
Newly acquired second mutations can confer drug resistance to Gleevec. These additional mutations that are not detected in the primary tumor often appear in new metastases of tumors being treated with Gleevec and in sections of otherwise responding tumors that start to grow (Chen et al, 2004; Antonescu et al, 2005; Wardelmann et al, 2005; Debiec-Rychter et al, 2005). Occasionally, resistant metastases may change enough to lose KIT-positivity, potentially mimicking other types of tumors (Pauwels et al, 2005). This means that pathologists must be alert to the possibility of disguised GISTs when evaluating new tumors in a GIST patient. Interestingly, it seems to be possible that additional mutations occur in later recurrences or metastases in the absence of any drug treatment (Vu et al, 2005).
As more drug options become available, mutation testing by pathologists will hopefully become more common in order to select the best drugs for the tumor’s characteristics, whether for primary tumors, metastases present at di
agnosis, or metastases developing during treatment.
More background information from the NIH in a “Guide to Understanding Genetic Conditions” can be found here: https://ghr.nlm.nih.gov/gene/KIT