

GIST in neurofibromatosis
Neurofibromatosis 1 (NF1) is a tumor suppressor gene encoding the protein neurofibromin, a member of the RAS regulatory protein family. Inactivation of the NF1 gene occurs in about 1 of 3000 births, making it a relatively common condition. NF1 inactivation can be a familial condition involving an autosomal dominant inheritance pattern. However, about half of cases may occur randomly (sporadic, not familial). Characteristics shown by NF1 patients include pigmentation abnormalities such as café au lait spots (light brown birthmarks), a predisposition to various cancers, and multiple neurofibromas (benign nerve sheath tumors). About 10% of patients develop malignant peripheral nerve sheath tumors.
Persons affected by NF1 have a much greater probability of developing gastrointestinal stromal tumor (GIST) than the general population. The prevalence of GIST is about 5-25% in the NF1 population, with the higher estimates including symptom-free tumors diagnosed only at autopsy (Ghrist, 1963; Fuller et al, 1991; Giuly et al, 2003; Zoller et al, 1997). Symptomatic GISTs in NF1 patients are usually diagnosed at an earlier age than typical sporadic GIST patients: in adulthood but prior to age 50. NF1 patients often show multiple GISTs, and GISTs of the small intestines outnumber gastric GISTs (in contrast to the higher percentage of gastric tumors in sporadic GIST patients). Tsukuda et al (2007) reported using laparoscopic surgery to remove intestinal GISTs in a NF-1 patient.
Mutation Findings
GIST tumors in NF1 usually have not shown mutations in the c-kit gene or the PDGFRA gene; that is, they have normal or “wild type” KIT and PDGRFA genes. Papers reporting exceptions include:
- Yantiss et al (2004) described a patient with an exon 11 mutation.
- Cheng (2004) also reported one mutant case.
- Takazawa et al (2005) reported mutations in at least one tumor from 3 of 9 NF1 patients. Both KIT and PDGFRA mutations were identified. Different tumors from a single patient could show different mutations, and the same patient could have both GISTs with mutations and other tumors that were wild-type.
- Mussi et al (2008) found c-kit mutations in primary tumors of 3 of 28 NF1 GIST patients (both exon 11 and exon 9) and a PDGFRA mutation in one patient. Additionally, a secondary mutation in exon 17 of c-kit was found after imatinib (Gleevec) treatment in one patient whose primay tumor had been wild-type. These researchers hypothesize that the mutations found in a small percentage of NF1 GIST patients are not causal.
Pathology and Prognostic Findings
The GISTs in NF1 patients are usually multiple and usually found on the small intestine, though a few have been found on the stomach. Areas of hyperplasia of ICCs between GISTs in NF1 may be precursor lesions (Andersson et al, 2005). NF1 GISTs do generally stain positive for KIT protein (CD117), like most other GISTs. Compared to sporadic GISTs, NF1 GISTs are more likely to show S-100 reactivity (a marker of neural differentiation), entrapped myenteric nerves within the tumor, and skeinoid fibers within the tumor (Takazawa et al, 2005).
Yamamoto et al (2008) found that all GISTs from five NF1 patients did stain positive for PKC theta.
Although they may fall into any GIST risk category, NF1 GISTs usually show low cell proliferation (growth) indicators such as mitotic count or Ki-67 index (Andersson et al, 2005; Miettinen et al, 2006). NF1 GISTs rarely metastasize (Levy et al, 2004). Andersson et al (2005) reported follow-up for 9 NF1 patients who had surgery for GIST; none of the patients died of GIST, and 6 of 9 were well up to 32 years later. Miettinen et al (2006) described follow-up for 35 NF1 GIST patients, of whom only 5 died of metastatic disease. Mussi et al (2008) reported follow-up for 28 NF1 GIST patients, of whom 7 (25%) developed metastases. In both of the preceding two series of NF1 GIST patients, none of those with multiple GISTs developed metastatic disease.
Pathways Activated in NF1 GIST
Because these GISTs generally do not exhibit c-kit or PDGFR mutations, they are apparently driven by some uncertain alternative mechanism. When KIT or PDGFRA mutations are found, they may be a late event in the tumor’s development, not a necessary event. The factors causing GIST in NF1 have not yet been identified. Andersson et al (2005) discuss the possibility that NF1 inactivation could lead to constitutive activation of ras and increased MAP kinase signaling. Maertens et al (2006) showed that inactivation of neurofibromin was sufficient to hyper-activate the MAPK pathway, which is more important in NF1 GISTs than in sporadic GISTs.
Yamamoto et al (2008) found that 92% of GISTs from NF1 patients, as well as precursor hyperplasia of interstitial cells of Cajal, showed activation of the MAPK p44/42 pathway. Loss of heterozygosity for chromosome arms 14q and 22q was common in their cases: 87% for 14q and 42% for 22q. There are probably tumor suppressor genes on these chromosomes whose loss contributes to GIST growth, but they have not yet been identified.
Stewart et al (2007) found that GISTs from two NF1 patients were homozygous for the mutant NF1 allele, having lost the normal wild-type allele through mitotic recombination. They hypothesized that loss of heterozygosity through mitotic recombination provides a mechanism for GIST development in NF1 patients, as is the case for development of neurofibromas.
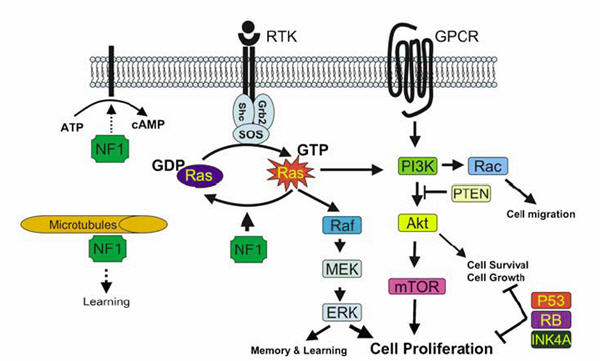
FIGURE 2 FROM LE AND PARADA (2007) IN ONCOGENE 26:4609-4616. REPRODUCED WITH PERMISSION OF NATURE PUBLISHING GROUP. REPRESENTATION OF NF1 INTERACTIONS WITH THE RAS AND PI3K PATHWAYS. NF1 CONSTRAINS RAS ACTIVITY IN THE NORMAL CELL. THEREFORE, LOSS OF NF1 EXPRESSION LEADS TO ELEVATED RAS ACTIVITY, DYSREGULATED CELL GROWTH AND TUMORIGENESIS. NF1 MAY ALSO ASSOCIATE WITH MICROTUBULES AND MODULATE THE CAMP-PKA SIGNALING PATHWAY.
Maertens et al (2006) showed that KIT activation in NF1 GIST cells depends on stem cell factor, in contrast to the constitutive KIT activation seen in sporadic GIST. Testing imatinib (Gleevec) in NF1 GIST cells, Maertens et al found that imatinib could not shut down the MAPK pathway, indicating that MAPK activation was not dependent on KIT or PDGFRA.
Other cell signaling pathways activated in NF1, as reviewed by Lee and Stephenson (2007) include adenylate cyclase > cAMP, mTOR, and protein kinase C.
There are no reports yet of the use of mTOR inhibitors or MAPK inhibitors on NF1 GIST cells or in NF1 patients with GIST. Johannssen et al (2008) found that MPNST cell lines from NF1 do show growth arrest (but not cell death) when exposed to rapamycin (an mTOR inhibitor). The rationale for use of mTOR and MAPK inhibitors in NF1 is clear.
Response of NF1 GIST to Imatinib and Sunitinib
There are only a few published papers about response of NF1 GISTs to tyrosine kinase inhibitor drugs:
- Lee et al (2006) reported a case of NF1 GIST that did respond to imatinib (Gleevec).
- Kalender et al (2007) reported a patient with initial response to imatinib (Gleevec) who subsequently became resistant and experienced progression. However, the metastatic lesions in liver and omentum did decrease in size during the first four cycles of sunitinib (Sutent). Results of longer follow-up were not provided in the paper.
- Mussi et al (2008) described imatinib treatment results for eight NF1 patients. Four patients who received adjuvant imatinib after complete resection did not experience recurrence. Four additional patients with metastases received imatinib, and three of them demonstrated primary resistance (rapid progression), while one patient with a PDGFRA mutation had stable disease temporarily.
Additional Resources
For an excellent slide show about GIST in NF1 patients, you can download the following Powerpoint presentation by Mussi et al from the 2007 CTOS program:
THERAPEUTIC CONSEQUENCES FROM MOLECULAR BIOLOGY FOR GIST PATIENTS AFFECTED BY NEUROFIBROMATOSIS TYPE 1
Chiara Mussi; Hans Ulrich Schildhaus; *Alessandro Gronchi; Eva Wardelmann; *Peter Hohenberger