

The Cell Cycle as a Drug Target
 GIST Support Internaional posed questions about new cancer drugs to Gary Schwartz, MD. Dr. Schwartz is chief of the Melanoma and Sarcoma Service in the Department of Medicine’s Division of Solid Tumor Oncology at Memorial Sloan-Kettering Cancer Center in New York. He is a medical oncologist who specializes in the identification and development of new targeted drugs for cancer therapy, particularly in the treatment of patients with gastrointestinal cancers and sarcomas. These agents are not disease-specific and hold promise in the treatment of all solid tumor malignancies because they affect cancer cells by targeting their progession through the cell cycle, the process resulting in proliferation and tumor growth.
GIST Support Internaional posed questions about new cancer drugs to Gary Schwartz, MD. Dr. Schwartz is chief of the Melanoma and Sarcoma Service in the Department of Medicine’s Division of Solid Tumor Oncology at Memorial Sloan-Kettering Cancer Center in New York. He is a medical oncologist who specializes in the identification and development of new targeted drugs for cancer therapy, particularly in the treatment of patients with gastrointestinal cancers and sarcomas. These agents are not disease-specific and hold promise in the treatment of all solid tumor malignancies because they affect cancer cells by targeting their progession through the cell cycle, the process resulting in proliferation and tumor growth.
1) Explain the function of histones to package DNA in a cell. How do histones affect gene expression?
Histones bind and package DNA. They cause chromatin structures to become compact and this restricts access to transcription factors, thus repressing the transcriptional expression of certain genes.
2) How are histones modified to fine tune their function? How do these modifications regulate gene expression?
Histones are modified by histone deacetylases (HDACs) by adding an acetyl group to the amino terminal lysine residue of the the histone. Decreased acetylation by HDACs cause the chromatin structures to become compact and this represses gene expression.
3) Explain the reversible dynamics of histone “acetylation and deacetylation?” How does this create a plasticity for gene expression?
There is a dynamic interaction between HDACs that removal acetyl groups and HATs (histone acetyl transferases) that add them. It is the addition of acetyl groups to the histones that results in chromatin uncoiling and gene transcription.
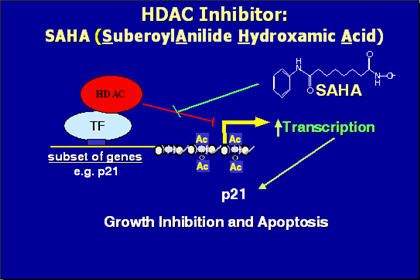
4) What are histone deacetylase inhibitors? How can these inhibitors stop cancer cells from growing?
HDAC inhibitors block the removal of acetyl groups by HDACs and tips the balance in favor of the HATs which increase the histone acetylation. This results in the transcriptional activation of specific genes (ie p21 or p27) that induce growth arrest by inactivating cyclin dependent kinases (CDKs). Proapoptotic genes may also be activated.
5) What is the cell cycle, and why is it important in cancer?
The cell cycle represents a series of events that the cell must pass through in order to grow and proliferate. The cell cycle has 4 phases: G1, S. G2, M. Each phase of the cell cycle has important machinery that the cell must pass through in order to survive. The transition points between the phases of the cell cycle (G1 to S or G2 to M) are regulated by CDKs, each CDK is regulated by positive factors (cyclins) and negative factors (cyclin dependent kinase inhibitors, CDKI, including p21 and p27). The purpose of the cell cycle is to recognize a cell with damaged DNA (such as a tumor cell or a tumor cell damaged by chemotherapy or radiation) and then trigger mechanisms that result in the eradication of the cell (a process of cell death called apoptosis). This process of self-regulation prevents the perpetuation of a “damaged” or “abnormal” cell through the cell cycle.
6) What is the effect of flavopiridol on GIST cancer cells?
Flavopiridol is a cell cycle inhibitor. It was initially identified from a plant and is now synthesized in the laboratory. It in essence services as a chemical CDKI by substituting for p21 or p27 where these may not be functional. In addition, it inhibits a specific CDK, called CDK9, which regulates RNA polymerase II. By inhibiting RNA Pol II, flavopiridol blocks the transcription of molecules which are critical for tumor cell survival. For GIST, by this mechanism, flavopiridol will then suppress the expression of c-kit resulting in cell death.
7) How might flavopiridol and a histone deacetylase inhibitor work synergistically as a GIST therapy?
Flavopiridol works on the mechanism of RNA transcription by inhibiting a kinase (RNA pol II) which drives the transcription of DNA to RNA, in this case c-kit, whereas an HDAC inhibitor works at the level opening up transcription. At this point this could in fact be antagonistic rather than synergistic.
However, some HDAC inhibitors may also work to inhibit HSP90. HSP90 is a chaperone protein that protects kit from proteasomal degradation. By inhibiting HSP90, this class of drugs may expedite the degradation of kit protein. Thus, by using a drug that blocks kit at the transcriptional (RNA) level and also at the protein (translational) level, the two classes of drugs could be synergistic. This requires preclinical testing.
8) Could either flavopiridol or a histone deacetylase inhibitor potentially work together with imatinib (Gleevec) to increase GIST cell death rather than just preventing GIST cell proliferation?
Yes, but preclinical studies indicate that flavopiridol is such a potent inhibitor of kit, that Gleevec plus flavopiridol was no better than flavopiridol alone. In fact, flavopiridol was a much more potent inducer of cell death than imatinib in preclinical studies. The theoretical advantage of flavopiridol is that it will inhibit transcription of all c-kits regardless of their mutational status, and therefore should be active even in imatinib resistant cells.
9) Is a trial of flavopiridol for GIST being planned?
We have no specific flavo trial for patients with GIST at this time, but recently at MSKCC we have begun to place patients who fail imatinib or other KIT inhibitors on the open phase I trials of flavopiridol plus chemotherapy, especially flavopiridol with gemcitabine. The hope is to first prove activity in these phase I clinical trials, and then propose to the National Cancer Institute (which regulates these clinical studies) a GIST-specific phase II trial.
Further Reading
Link here for PubMed abstracts and/or full text of selected papers by Dr. Schwartz and colleagues about flavopiridol and SAHA, a histone deacetylase inhibitor.
Link to Marina Symcox’s tutorial Understanding How DNA Is Packaged in a Cell.